

We will perform quantum mechanics based molecular dynamics simulations (AIMD) for studying the electron transfer from electrode to molecules that can be used in the redox flow battery (RFB).

To be able to model the charge transfer we use the Constrained Density Functional Theory (cDFT). With cDFT we can force an electron to be either on the electrode (in our case a graphene sheet) or on the molecule. We need to run two parallel AIMD simulations with different charge localization and from these simulations we can compute parameters used in Marcus theory.
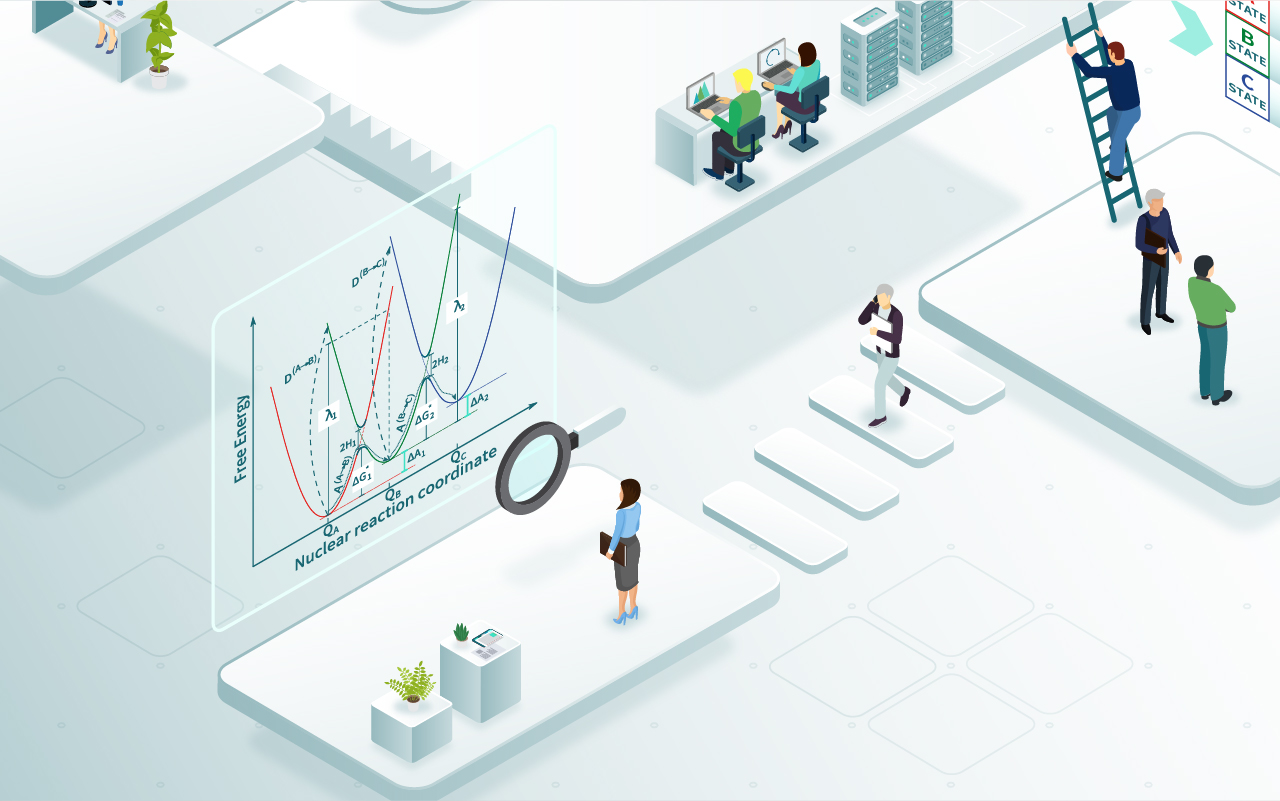

The Marcus theory will predict the electron transfer rate. This approach does not have any parameters that should be fitted to experiments so it will provide unbiased predictions of electrons transfer rates for particular molecules. The electrode to molecule charge transfer rate is an important property for RFB. If it is very slow the whole RFB can be inefficient. Unfortunately, this quantity is difficult to measure and difficult to simulate.

Our cDFT simulations are among the first this type of simulations in the world, and they will open a new way to study the electrode-molecule charge transfer. The first AIMD simulations have been quite time consuming but we are exploring more approximate but much faster ways to get the charge transfer rates.