

Electrochemical redox reactions kinetics modeling at the electrode-electrolyte interface
We use quantum mechanical constraint density functional theory (CDFT) in combination with ab initio molecular dynamics (AIMD) to model the kinetics of electrochemical redox reactions at the electrode-electrolyte interface. Marcus theory is used to couple two states of reactant and product with electron transfer kinetics. We also study the resulting kinetics of reactions between some organic redox active molecules as anolyte and graphene as electrode. All these molecules undergo consecutive electrochemical reactions with one electron being transferred at each stage. This theoretical study contributes towards the development of a realistic-level prediction of electron transfer kinetics suitable for energy storage applications.

LUMI supercomputer used in the simulations.
Aalto preliminary results
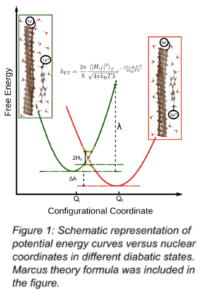 By introducing a two-state system of reactant (i) and product (f), Marcus theory (MT) estimates the interfacial electron transfer (ET) rate constant, which is a key parameter for analyzing the kinetic reactions during the charging and discharging processes of RFBs. All parameters of the theory (see FIG 1) are calculated using the quantum mechanical constraint density functional theory method in combination with ab initio molecular dynamics. We model the electrode with a single layer of graphene and Methyl-viologen (Me-Vi), 6,6’-dimethyldiquat (DMDQ), desalted Basic Red (dBR5), 2-hydroxy-1,4-naphthaquinone (2HNQ), and 1,1’-di(2-ethanol)-4,4’-bipyridinium (OH-Vi) were selected for the electrolyte molecules.
By introducing a two-state system of reactant (i) and product (f), Marcus theory (MT) estimates the interfacial electron transfer (ET) rate constant, which is a key parameter for analyzing the kinetic reactions during the charging and discharging processes of RFBs. All parameters of the theory (see FIG 1) are calculated using the quantum mechanical constraint density functional theory method in combination with ab initio molecular dynamics. We model the electrode with a single layer of graphene and Methyl-viologen (Me-Vi), 6,6’-dimethyldiquat (DMDQ), desalted Basic Red (dBR5), 2-hydroxy-1,4-naphthaquinone (2HNQ), and 1,1’-di(2-ethanol)-4,4’-bipyridinium (OH-Vi) were selected for the electrolyte molecules.
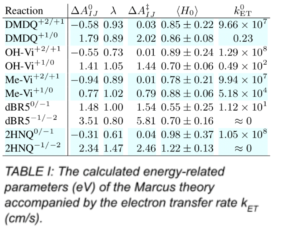
The kinetics of reactions can be understood using the MT parameters listed in TABLE I. In most cases, the transfer rate is high, which is not quantitatively consistent with experiments. In general, two types of reactions are expected on the surface of the electrode. First, the molecule comes close enough to accept an electron and, in the best case, leaves the interface. Second, the molecule sticks to the surface and poisons the catalytic activity of the electrode, which is unfavorable. While the former is called an outer-sphere ET, the latter is an inner-sphere with a much higher transfer rate. Our results show that both types are possible in RFBs, depending on the nature of the components. For example, we found that dicationic viologen-based molecules are strongly adsorbed when the first electron is embarked. At the smallest distance, we expect an inner-sphere ET, which is also thermodynamically favorable.
This leads us to rethink about the realistic electrode: the electrode surface could be covered by the first few redox active molecules. This may prevent next molecules to stick to the surface. We are now investigating the distance-dependent ÉT on clean and molecule covered surface to see if we can find the outer-sphere reaction for viologen-based molecules.
As another example, we would like to mention, is the phenazine-based molecule. The mechanism is different because of the protonation that must be included at some stage. Our results for a pH neutral electrolyte show that both ETs are an outer-sphere with two well separated reaction potentials. We think that the second ET is probably inaccessible unless the pH is changed. Our molecular-level computational analyses and calculations can be used to study ET in detail, paving the way for future screening of larger data sets.
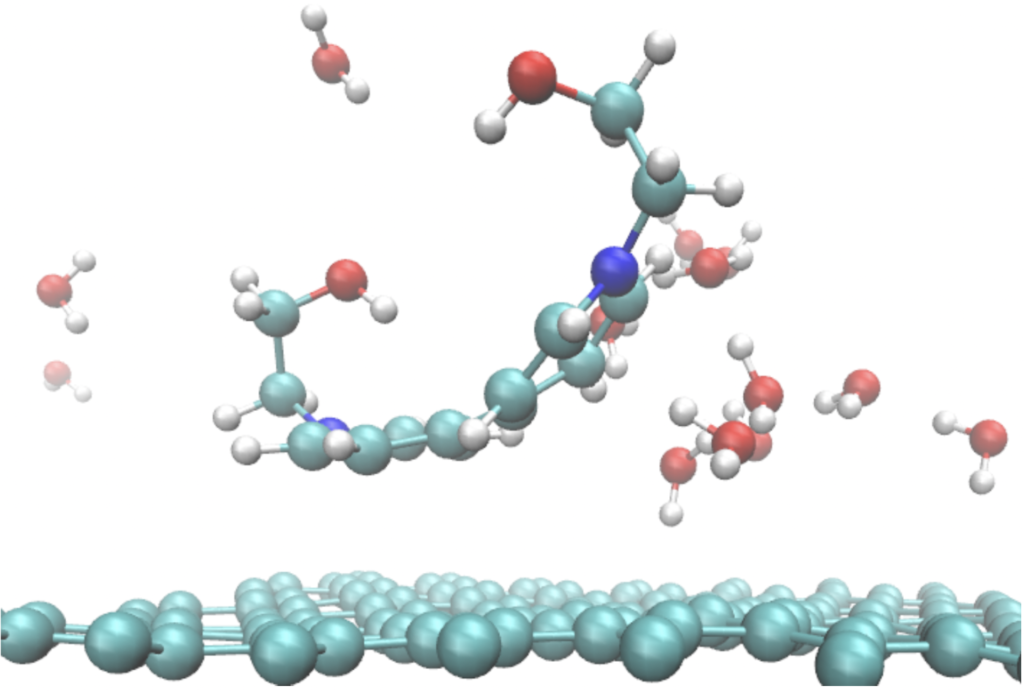
Snapshot of a simulation the OH-Viologen molecule on graphene.